# 蛋白质生物学的世界模型:ESMC、ESMFold2 与 ESM Atlas
[ESMC 与 ESMFold2 预印本](https://www.biorxiv.org/content/10.64898/2026.06.03.729735) ⋅ [Atlas](https://biohub.ai/esm/protein/atlas) ⋅ [教程](https://github.com/Biohub/esm/tree/main/cookbook/tutorials) ⋅ [Slack](https://bit.ly/esm-slack)
我们正在发布一个针对蛋白质生物学的世界模型:一个用于预测、设计和发现的前沿科学引擎。该系统建立在最新一代的进化尺度建模(Evolutionary Scale Modeling, ESM)基础之上,从进化产生的蛋白质序列中学习,并利用这些知识在各种尺度上表征、映射、预测和设计蛋白质——从原子间相互作用到跨越数十亿年的进化关系。该系统包含三个核心产出:ESMC、ESMFold2 和 ESM Atlas。
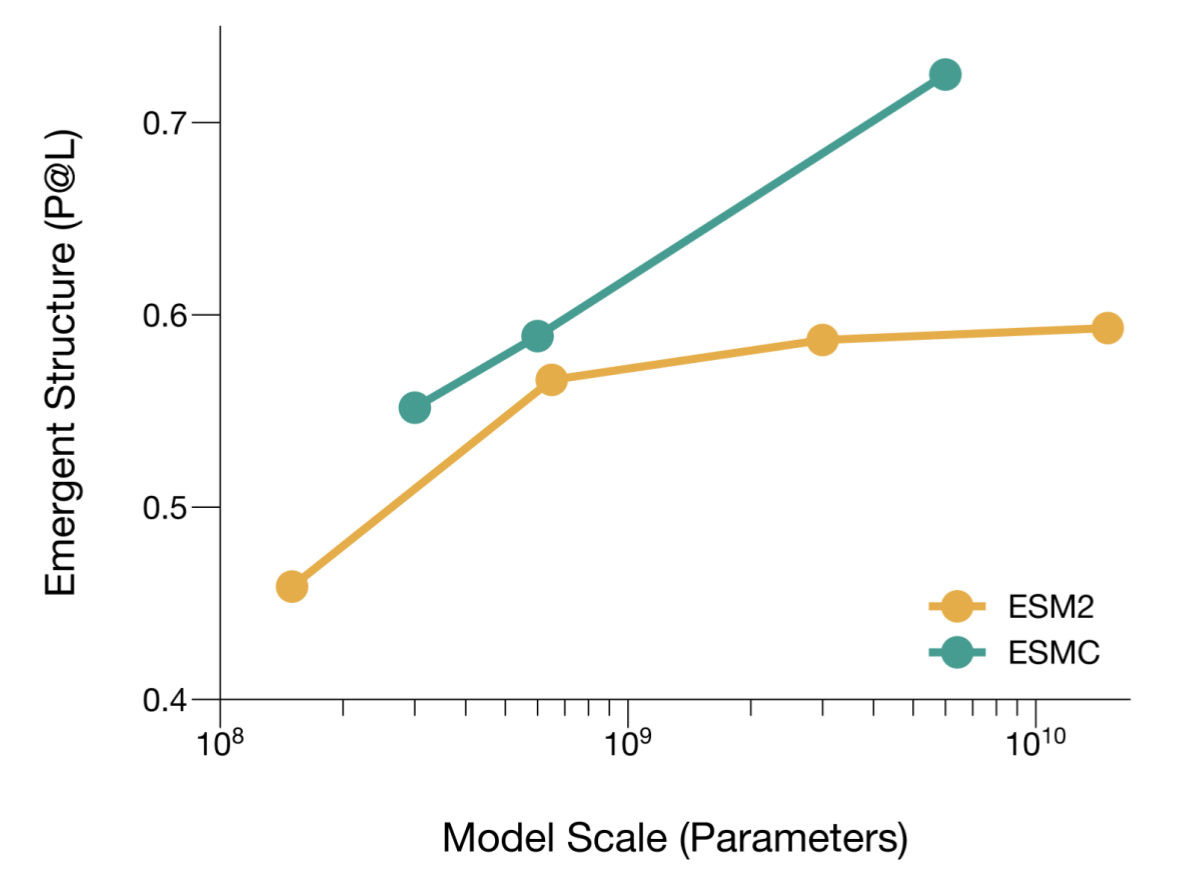
**[ESMC](https://biohub.ai/esm/protein)** 是一个最先进的蛋白质语言模型,它通过在数十亿蛋白质序列上进行训练,掌握了蛋白质生物学的规律。与 ESM2 相比,ESMC 定义了一个新的扩展前沿,随着模型规模的增加,在涌现出的长距离结构理解方面实现了更强大的性能。
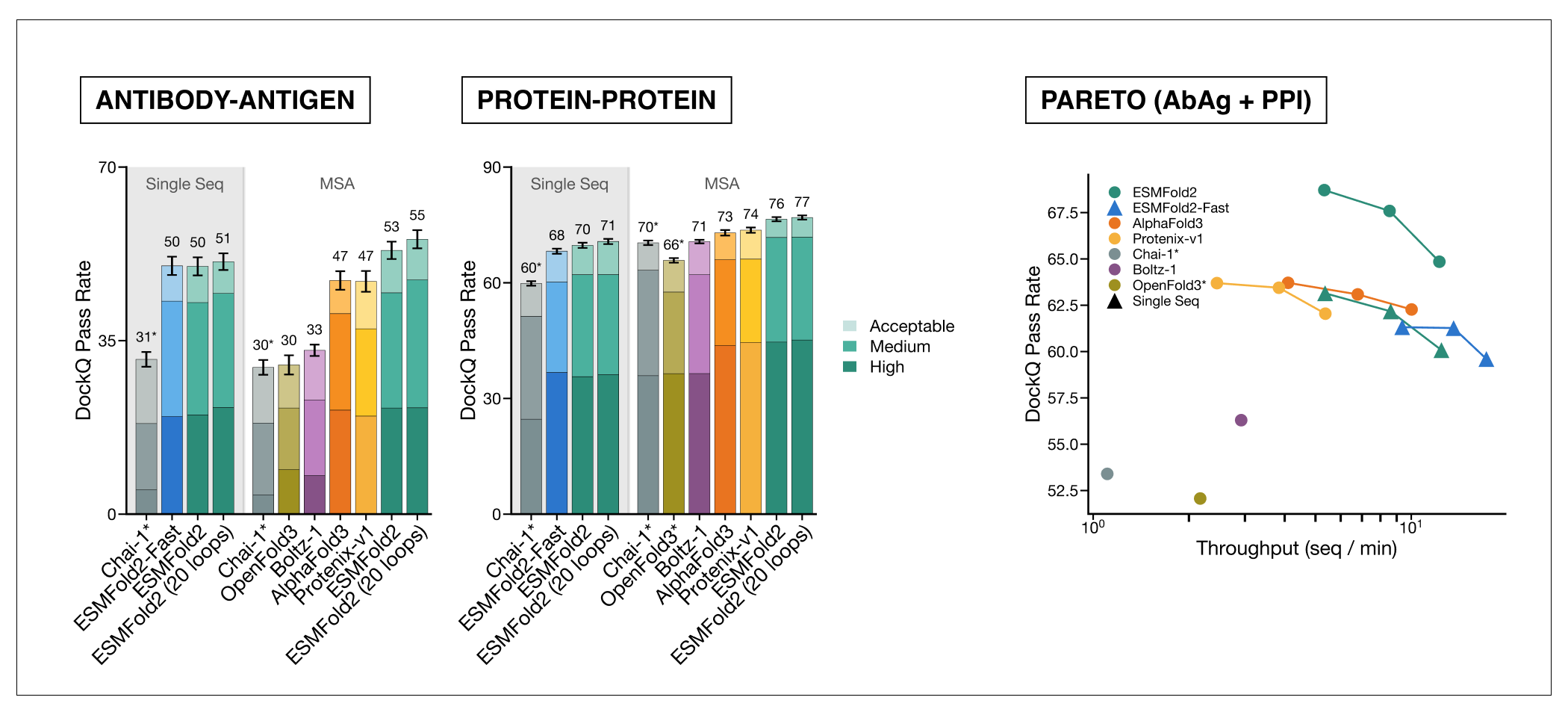
**[ESMFold2](https://huggingface.co/biohub/ESMFold2)** 基于 ESMC 6B 模型构建,是一个最先进的结构预测模型,已在蛋白质-蛋白质相互作用设计方面得到验证。在 Foldbench 蛋白质-蛋白质及抗体-抗原复合物的 DockQ 通过率上,ESMFold2 超越了其他模型,并且可以在单序列模式下运行,从而将折叠速度提高一个数量级。
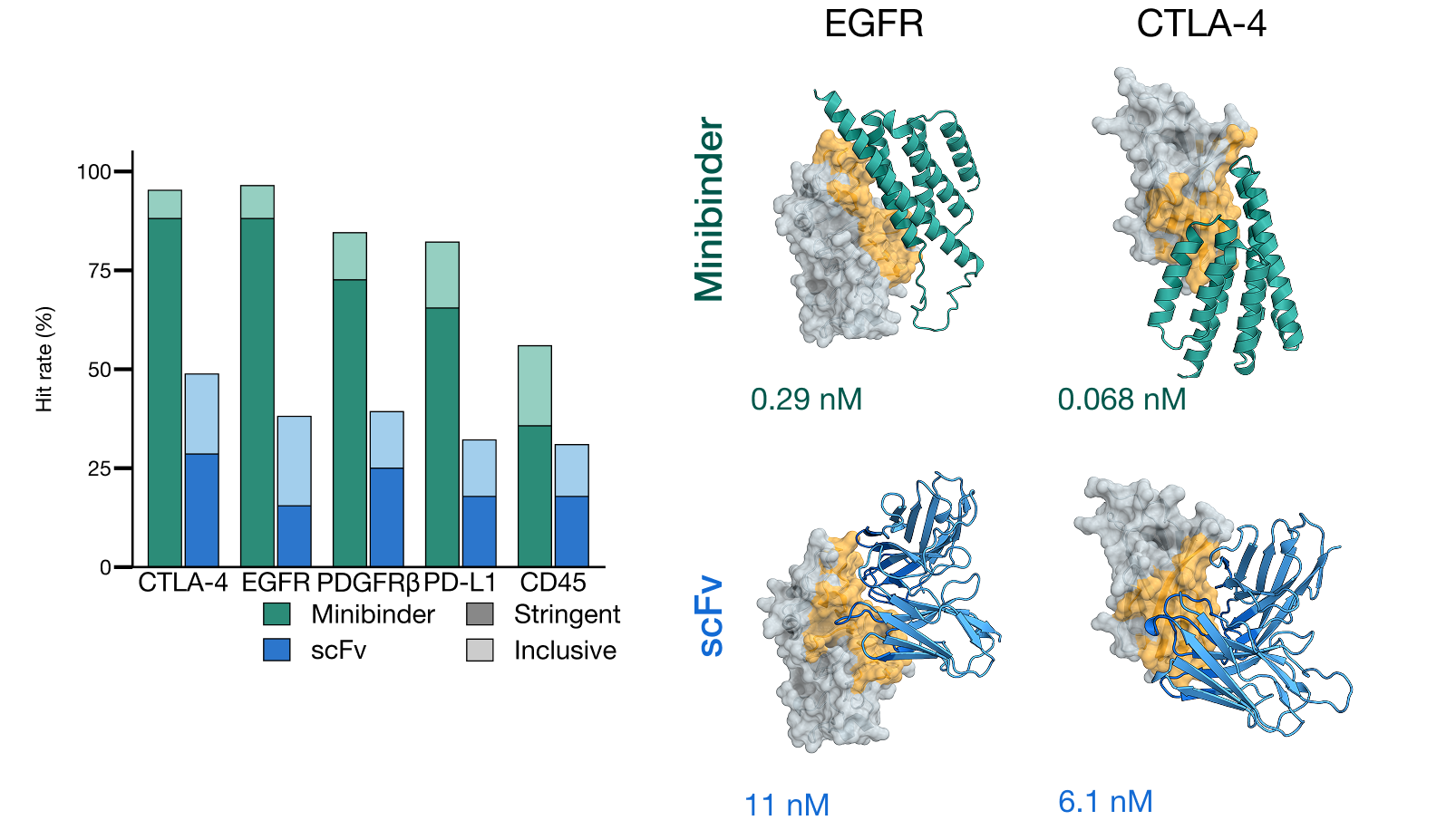
ESMFold2 已在实验室中跨五个治疗靶点进行了验证。ESMFold2 的反演(Inversion)功能使得能够从头生成具有高命中率、纳摩尔级亲和力、靶向特异性和功能活性的微型结合剂和源自抗体的 scFv。我们在这个[笔记本](https://github.com/Biohub/esm/blob/main/cookbook/tutorials/binder_design.ipynb)中发布了从目标序列到排序结合剂设计的完整协议。有关更多详细信息,请参阅[预印本](https://www.biorxiv.org/content/10.64898/2026.06.03.729735)。
**[ESM Atlas](https://biohub.ai/esm/protein/atlas)** 是一张涵盖 68 亿个蛋白质的地图,包含了生命生物多样性的全部广度。ESMFold2 的折叠吞吐量使得预测超过十亿个结构成为可能。该 Atlas 根据 ESMC 的内部世界模型进行组织。我们通过训练稀疏自编码器使这个世界模型具有可解释性。SAE 是一种无监督神经网络,经过训练可以将 ESMC 的内部表征分解为大约 16,000 个稀疏的、可解释的特征,从而揭示 ESMC 所学习到的蛋白质之间的功能关系。每个特征都通过智能体流水线用自然语言进行了总结,该流水线将特征映射到蛋白质数据库中已知的生物学知识。我们发布了一系列在不同模型规模、不同层以及不同粒度级别上训练的 SAE。请在 [Biohub 平台](https://biohub.ai/)上了解更多关于如何使用 ESM Atlas 的信息。
有关使用 ESM3 的信息,请参阅 [ESM3 README](https://github.com/Biohub/esm/blob/main/_assets/ESM3_README.md)。
## 目录
- [ESMC](#esmc)
- [ESMC 稀疏自编码器](#esmc-sparse-autoencoders)
- [ESMFold2](#esmfold2)
- [前沿安全](#frontier-safety)
- [许可证](#licenses)
- [引用](#citations)
## ESMC
[ESMC](https://biohub.ai/esm/protein) 是一个最先进的蛋白质语言模型,它通过在数十亿蛋白质序列上进行训练,学习到了蛋白质生物学的表征。
ESMC 的代码库、模型权重和模型变体可通过 [Hugging Face](https://huggingface.co/collections/biohub/esmc-model-family) 获取。
运行 ESM 模型主要有两种方式:通过 [**Biohub 平台**](https://biohub.ai/) 或在本地使用 Hugging Face。Biohub 平台使用户能够以最少的设置轻松运行 ESM 模型进行推理。有兴趣自定义或微调 ESM 模型的用户可以使用来自 Hugging Face 的模型。
### 通过 Hugging Face 运行 ESMC
首先,从 GitHub 安装 `esm`(PyPI 版本即将推出):
```
pip install esm@git+https://github.com/Biohub/esm.git@main
```
然后使用以下代码,通过 Hugging Face 的 Transformers 库运行 ESMC:
```
import torch
from transformers import AutoModelForMaskedLM, AutoTokenizer
from huggingface_hub import login
# 使用您的 Hugging Face 凭据登录
login()
# GFP 序列示例
sequences = ["MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK"]
model = AutoModelForMaskedLM.from_pretrained(
"biohub/ESMC-6B",
device_map="auto",
).eval()
tokenizer = AutoTokenizer.from_pretrained("biohub/ESMC-6B")
inputs = tokenizer(sequences, return_tensors="pt", padding=True)
inputs = {k: v.to(model.device) for k, v in inputs.items()}
with torch.inference_mode():
output = model(**inputs)
```
默认情况下,模型仅返回最后一层的表征。要返回**所有 transformer 层**的隐藏状态,请设置:
```
output = model(**inputs, output_hidden_states=True)
```
### 通过 Biohub 平台运行 ESMC
以下代码展示了如何使用 Biohub 平台访问 ESMC。可以在[开发者控制台](https://biohub.ai/developer-console/api-keys)中创建 API 令牌。
请注意,我们的 API 已从 forge.evolutionaryscale.ai 迁移至 [biohub.ai](https://biohub.ai),因此某些代码类中仍引用了“Forge”。
要开始使用 ESM,请使用 `pip` 安装该 Python 库:
```
pip install esm@git+https://github.com/Biohub/esm.git@main
```
然后导入必要的库并实例化您所需的模型。
```
from esm.sdk import esmc_client
from esm.sdk.api import ESMProtein, LogitsConfig
# Human carbonic anhydrase II (PDB 2CBA)
protein = ESMProtein(
sequence=(
"MSHHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDD"
"SQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVL"
"GIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEP"
"ISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK"
)
)
model = esmc_client(
model="esmc-600m-2024-12", url="https://biohub.ai", token="
"
)
protein_tensor = model.encode(protein)
logits_output = model.logits(
protein_tensor, LogitsConfig(sequence=True, return_embeddings=True)
)
print(logits_output.logits, logits_output.embeddings)
```
有关如何使用 ESMC 的教程,请参阅我们的[教程](https://github.com/Biohub/esm/tree/main/cookbook/tutorials)。
## ESMC 稀疏自编码器 (SAE)
稀疏自编码器(SAE)是一种无监督方法,用于将大型 transformer 语言模型的表征分解为可解释的单元。我们发布了在 ESMC 上训练的 SAE,以揭示 ESMC 世界模型所学习到的、可解释的功能组织单元。
Atlas 中使用并在论文中进行分析的稀疏自编码器 `ESMC-6B-sae-layer60-k64-codebook16384` 是基于 ESMC 6B 模型构建的。我们还为此 SAE 的码本提供了人类易于理解的、由智能体生成的特征描述。
ESMC SAE 的代码库、模型权重和模型变体可通过 [Hugging Face](https://huggingface.co/collections/biohub/esmc-saes-for-hidden-states-all-layers) 获取。
### 通过 Hugging Face 运行 SAE
首先,从 GitHub 安装 `esm`(PyPI 版本即将推出):
```
pip install esm@git+https://github.com/Biohub/esm.git@main
```
然后使用以下代码,通过 Hugging Face 的 Transformers 库设置 ESMC SAE:
```
import torch
from transformers import AutoModel, AutoTokenizer
sequence = "MGSNKSKPKDASQRRRSLEPAENVHGAGGGAFPASQTPSKPASADGHRGPSAAFAPAAAEPKLFGGFNSSDTVTSPQRAGPLAGGVTTFVALYDYESRTETDLSFKKGERLQIVNNTEGDWWLAHSLSTGQTGYIPSNYVAPSDSIQAEEWYFGKITRRESERLLLNAENPRGTFLVRESETTKGAYCLSVSDFDNAKGLNVKHYKIRKLDSGGFYITSRTQFNSLQQLVAYYSKHADGLCHRLTTVCPTSKPQTQGLAKDAWEIPRESLRLEVKLGQGCFGEVWMGTWNGTTRVAIKTLKPGTMSPEAFLQEAQVMKKLRHEKLVQLYAVVSEEPIYIVTEYMSKGSLLDFLKGETGKYLRLPQLVDMAAQIASGMAYVERMNYVHRDLRAANILVGENLVCKVADFGLARLIEDNEYTARQGAKFPIKWTAPEAALYGRFTIKSDVWSFGILLTELTTKGRVPYPGMVNREVLDQVERGYRMPCPPECPESLHDLMCQCWRKEPEERPTFEYLQAFLEDYFTSTEPQYQPGENL"
model = AutoModel.from_pretrained("biohub/ESMC-6B", device_map="auto").eval()
tokenizer = AutoTokenizer.from_pretrained("biohub/ESMC-6B")
sae = AutoModel.from_pretrained(
"biohub/ESMC-6B-sae-k64-codebook16384",
allow_patterns=["config.json", "layer_30.safetensors", "layer_60.safetensors"],
device=model.device,
)
sae.initialize_layers([30, 60])
model.add_sae_models([sae.layers["30"], sae.layers["60"]])
inputs = tokenizer(sequence, return_tensors="pt", padding=True)
inputs = {k: v.to(model.device) for k, v in inputs.items()}
with torch.inference_mode():
output = model(**inputs)
output["sae_outputs"]["layer60"] # sparse.coo tensor
print(output["sae_outputs"]["layer60"].shape)
```
### 通过 Biohub 平台运行 SAE
有关使用 Biohub 平台运行 SAE 的教程,请参见[此处](https://github.com/Biohub/esm/blob/main/cookbook/tutorials/esmc_sae_feature_interpretation.ipynb)。
## ESMFold2
[ESMFold2](https://huggingface.co/biohub/ESMFold2) 是一个最先进的蛋白质结构预测模型,它将 ESMC(60 亿参数)语言模型的嵌入与基于扩散的结构预测架构相结合。
该模型直接从氨基酸序列预测高分辨率的全原子 3D 蛋白质结构,并可选地输入多序列比对(MSA),以提高在挑战性目标上的准确性。ESMFold2 在多样化的评估数据集上取得了匹配甚至超越 AlphaFold3 的最先进性能,同时通过优化的扩散采样和架构创新提供了更高的计算效率。
ESMFold2 的代码库、模型权重和模型变体可通过 [Hugging Face](https://huggingface.co/biohub/ESMFold2) 获取。
### 通过 Hugging Face 运行 ESMFold2
首先,从 GitHub 安装 `esm`(PyPI 版本即将推出):
```
pip install esm@git+https://github.com/Biohub/esm.git@main
```
然后使用以下代码,通过 Hugging Face 的 Transformers 库在本地运行 ESMFold2:
```
from esm.models.esmfold2 import (
DNAInput,
ESMFold2InputBuilder,
LigandInput,
Modification,
ProteinInput,
StructurePredictionInput,
)
from transformers.models.esmfold2.modeling_esmfold2 import ESMFold2Model
HHAI_SEQ = (
"MIEIKDKQLTGLRFIDLFAGLGGFRLALESCGAECVYSNEWDKYAQEVYEMNFGEKPEGDITQVNEKTIPDH"
"DILCAGFPCQAFSISGKQKGFEDSRGTLFFDIARIVREKKPKVVFMENVKNFASHDNGNTLEVVKNTMNELD"
"YSFHAKVLNALDYGIPQKRERIYMICFRNDLNIQNFQFPKPFELNTFVKDLLLPDSEVEHLVIDRKDLVMTN"
"QEIEQTTPKTVRLGIVGKGGQGERIYSTRGIAITLSAYGGGIFAKTGGYLVNGKTRKLHPRECARVMGYPDS"
"YKVHPSTSQAYKQFGNSVVINVLQYIAYNIGSSLNFKPY"
)
model = ESMFold2Model.from_pretrained("biohub/ESMFold2").cuda().eval()
spi = StructurePredictionInput(
sequences=[
ProteinInput(id="A", sequence=HHAI_SEQ),
DNAInput(
id="B",
sequence="GATAGCGCTATC",
modifications=[Modification(position=5, ccd="C36")],
),
DNAInput(
id="C",
sequence="TGATAGCGCTATC",
modifications=[Modification(position=6, ccd="C36")],
),
LigandInput(id="L", ccd=["SAH"]),
]
)
result = ESMFold2InputBuilder().fold(
model, spi, num_loops=20, num_sampling_steps=100, num_diffusion_samples=1, seed=0
)
print(f"pLDDT mean: {float(result.plddt.mean()):.3f}, pTM: {float(result.ptm):.3f}, ipTM: {float(result.iptm):.3f}")
with open("1mht_pred.cif", "w") as f:
f.write(result.complex.to_mmcif())
```
### 通过 Biohub 平台运行 ESMFold2
安装 `esm` Python 包
```
pip install esm@git+https://github.com/Biohub/esm.git@main
```
导入必要的库。
```
from esm.sdk.forge import SequenceStructureForgeInferenceClient
from esm.sdk.api import FoldingConfig
from esm.utils.structure.input_builder import ProteinInput, StructurePredictionInput
```
调用带有选定模型的推理客户端,并将 替换为您的令牌名称。
```
client = SequenceStructureForgeInferenceClient(model="esmfold2-fast-2026-05", url="https://biohub.ai", token="")
# Human carbonic anhydrase II (PDB 2CBA)
ca2_sequence = (
"MSHHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDD"
"SQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVL"
"GIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEP"
"ISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK"
)
ca2_input = StructurePredictionInput(
sequences=[ProteinInput(id="A", sequence=ca2_sequence)]
)
config = FoldingConfig(
num_loops=20,
num_sampling_steps=100
)
result = client.fold_all_atom(ca2_input, config=config)
with open("result.cif", "w") as f:
f.write(result.complex.to_mmcif())
```
有关如何使用 ESMFold2 的教程,请参阅我们的[教程](https://github.com/Biohub/esm/tree/main/cookbook/tutorials)。
## 前沿安全
评估:在发布之前,我们进行了评估,以增进我们对特定与滥用相关的功能任务的能力提升的理解。这些评估的完整详细信息可在我们对应论文的附录中找到。
Biohub 平台:我们在免费开放的平台上实施了防护措施,以检测和限制使用与受控病原体和毒素相关的关键词及序列。有关这些防护措施的更多详细信息,请参阅我们的 Biohub 平台资源页面。我们认识到,使用 AI 模型来理解和建模这些序列和蛋白质有许多正当理由。如果您的研究工作受到这些防护措施的影响,您可以通过 [biohub.ai](https://biohub.ai) 申请提升我们平台的访问权限。
## 许可证
这些模型基于 [MIT 许可证](https://github.com/Biohub/esm/blob/main/LICENSE.md)提供。
## 引用
如果您的工作中使用了 ESM,请引用以下内容之一:
#### ESMC、SAE 和 ESMFold2
```
@misc{candido2026language,
title = {Language Modeling Materializes a World Model of Protein Biology},
author = {Candido, Salvatore and Hayes, Thomas and Derry, Alexander and Rao, Roshan
and Lin, Zeming and Verkuil, Robert and Wu, Bryan and Lee, Jin Sub
and Bruguera, Elise S. and Keval, Jehan A. and Kopylov, Mykhailo
and Pak, John E. and Wu, Wesley and Thomas, Neil and Mataraso, Samson
and Hsu, Alvin and Trotman-Grant, Ashton C. and Fatras, Kilian
and dos Santos Costa, Allan and Badkundri, Rohil and Ak{\i}n, Halil
and Oktay, Deniz and Deaton, Jonathan and Montabana, Elizabeth
and Sitwala, Hrishita and Yu, Yue and Wiggert, Marius
and Carlin, Dylan Alexander and Goering, Anthony W. and Blazejewski, Tomasz
and Sandora, McCullen and Hla, Michael and Jia, Tina Z.
and Kloker, Leon H. and Sofroniew, Nicholas J. and Uehara, Masatoshi
and Pannu, Jassi and Bachas, Sharrol and Liu, Daniel S.
and Sercu, Tom and Rives, Alexander},
year = {2026},
url = {https://www.biorxiv.org/content/10.64898/2026.06.03.729735},
note = {Preprint}
}
```
#### ESM3
```
@article {hayes2024simulating,
author = {Hayes, Thomas and Rao, Roshan and Akin, Halil and Sofroniew, Nicholas J. and Oktay, Deniz and Lin, Zeming and Verkuil, Robert and Tran, Vincent Q. and Deaton, Jonathan and Wiggert, Marius and Badkundri, Rohil and Shafkat, Irhum and Gong, Jun and Derry, Alexander and Molina, Raul S. and Thomas, Neil and Khan, Yousuf A. and Mishra, Chetan and Kim, Carolyn and Bartie, Liam J. and Nemeth, Matthew and Hsu, Patrick D. and Sercu, Tom and Candido, Salvatore and Rives, Alexander},
title = {Simulating 500 million years of evolution with a language model},
year = {2025},
doi = {10.1126/science.ads0018},
URL = {http://dx.doi.org/10.1126/science.ads0018},
journal = {Science}
}
```
#### ESM Github(代码 / 权重)
```
@software{evolutionaryscale_2024,
author = {{EvolutionaryScale Team}},
title = {evolutionaryscale/esm},
year = {2024},
publisher = {Zenodo},
doi = {10.5281/zenodo.14219303},
URL = {https://doi.org/10.5281/zenodo.14219303}
}
```