Dorif/fragalyseqt
GitHub: Dorif/fragalyseqt
跨平台开源的 DNA 片段分析数据处理软件,用于毛细管电泳数据的峰检测、sizing、等位基因分箱及 CODIS 格式导出。
Stars: 26 | Forks: 4
# README
 ## 这是什么?
这是一款用于片段分析(MLPA、QF-PCR 等)数据处理的软件。
## 这是什么?
这是一款用于片段分析(MLPA、QF-PCR 等)数据处理的软件。
 ## 功能特性
FragalyseQt 0.4 及以上版本支持同时使用多达 8 种不同染料的检测,支持选择性通道隐藏、非拉丁语运行名称,能够正确处理损坏的文件,支持为任何 FSA 或 HID 文件以 CSV 格式导出峰位置、面积、FWHM、大小和通道名称,并支持将 ABI 3500 和 SeqStudio 系列设备生成的 FSA 或 HID 文件的内部分析数据导出为 CSV 格式。自 0.4.2 版本起,FragalyseQt 还支持 Global 和 Local Southern sizing(如 doi: 10.1016/0003-2697(79)90235-5 中所述)。峰面积的计算假设其符合高斯分布。对于噪声较大的运行,提供了去噪和基线校正选项。
自 0.5 版本起,支持使用来自 GeneMapper(制表符分隔的 `*_Panel*.txt`,可附带 `*_Bins*.txt` 和 `*_Stutter*.txt`)和 GeneMarker (`.xml`) 的 panel 文件进行等位基因分箱。**NCBI OSIRIS LadderInfo XML** panel 文件(`.xml`,根元素为 `
## 功能特性
FragalyseQt 0.4 及以上版本支持同时使用多达 8 种不同染料的检测,支持选择性通道隐藏、非拉丁语运行名称,能够正确处理损坏的文件,支持为任何 FSA 或 HID 文件以 CSV 格式导出峰位置、面积、FWHM、大小和通道名称,并支持将 ABI 3500 和 SeqStudio 系列设备生成的 FSA 或 HID 文件的内部分析数据导出为 CSV 格式。自 0.4.2 版本起,FragalyseQt 还支持 Global 和 Local Southern sizing(如 doi: 10.1016/0003-2697(79)90235-5 中所述)。峰面积的计算假设其符合高斯分布。对于噪声较大的运行,提供了去噪和基线校正选项。
自 0.5 版本起,支持使用来自 GeneMapper(制表符分隔的 `*_Panel*.txt`,可附带 `*_Bins*.txt` 和 `*_Stutter*.txt`)和 GeneMarker (`.xml`) 的 panel 文件进行等位基因分箱。**NCBI OSIRIS LadderInfo XML** panel 文件(`.xml`,根元素为 ``)也同样受支持。
加载 panel 后,已确定大小的峰将自动分配等位基因名称;不在任何已知 bin 内的峰将被标记为 OL(off ladder,脱梯)。当加载了 GeneMapper stutter 文件或带有 stutter 比例的 GeneMarker panel 时,将自动应用 **stutter 峰过滤**:高度低于 panel 定义的 stutter 比例(相对于相邻等位基因)的峰将被标记为 stutter 产物。请注意,NCBI OSIRIS panel 不包含 stutter 比例数据,因此不适用于它们进行 stutter 过滤。同样自 0.5 版本起,支持导出为 **CODIS 3.2 CMF XML** 格式:使用 `Ctrl+Shift+C` 打开导出对话框,选择要包含的样本,为每个样本分配标本类别和 ID,填写实验室元数据,FragalyseQt 将生成一个 Schema 有效的 `CODISImportFile`,随时可提交至数据库。所有 29 个 CODIS 标本类别均受支持;基因座数据从已加载的 panel 分配中自动提取。
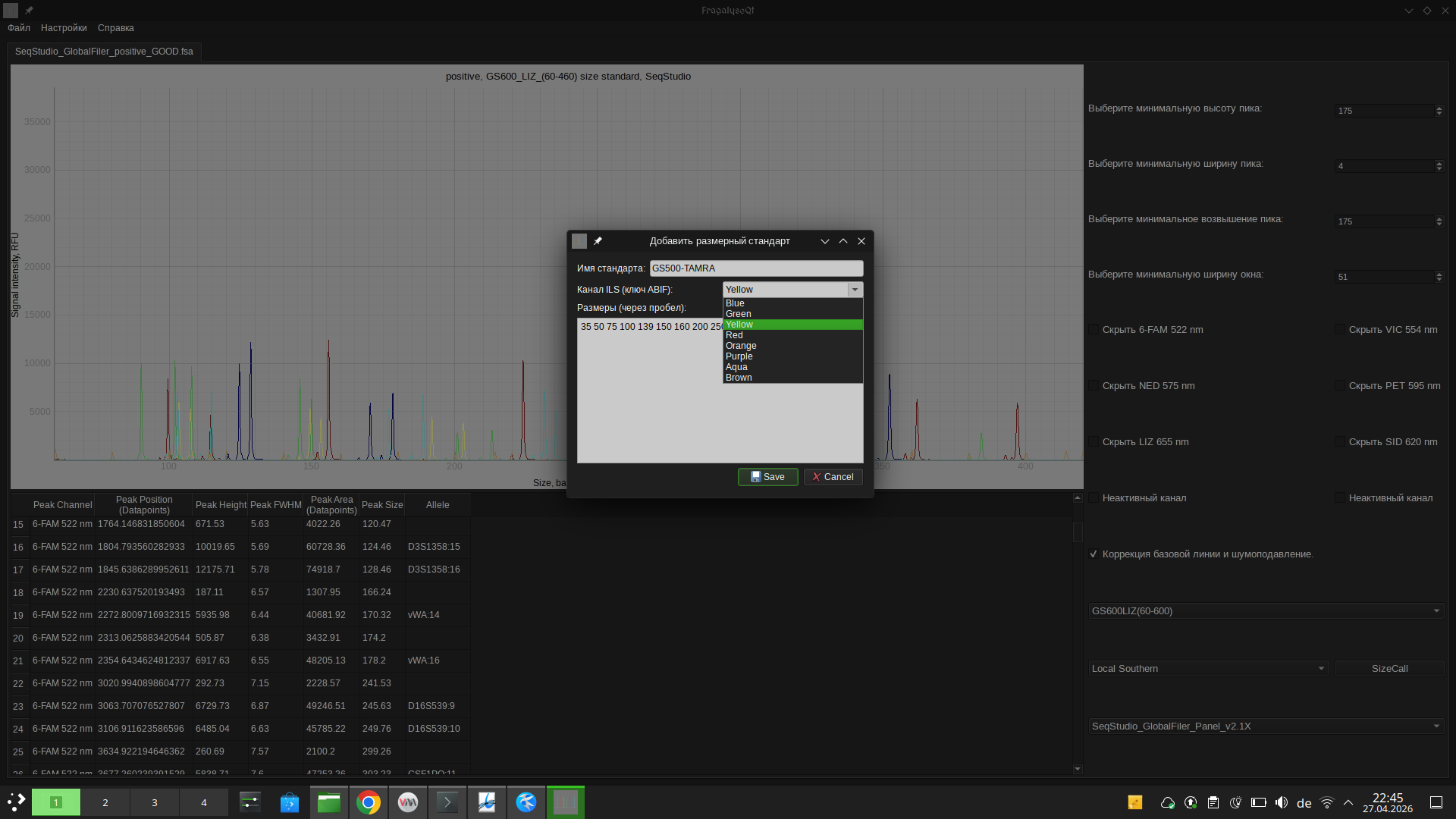
可以使用 **添加分子量内标** 对话框 (`Ctrl+Shift+A`) 将自定义分子量标准添加到库中:输入名称,选择 ILS 通道(DATA1-4 对应 Blue/Green/Yellow/Red,扩展通道 DATA105-108 对应 Orange/Purple/Aqua/Brown),并提供以空格分隔的大小列表。新标准将被永久保存,并适用于所有当前打开和未来的分析标签页。
所有这些操作都不会改写 FragalyseQt 所处理文件内部的任何数据!
## 支持哪些文件格式?
目前支持 FSA 和 HID 文件,并且(自 0.4.3 版本起)增加了针对原生 Nanophore-05 FRF 文件的初步实验性支持(不适用于 sizing,仅用于查看)。
如果您希望增加对任何文件格式的支持,可以通过发送补丁来协助实现。
## FragalyseQt 有什么用处?
目前,它有助于毛细管电泳的故障排除、峰检测和 sizing,因此适用于大量不同的分析工作。
由于 FragalyseQt 分析的是原始的毛细管电泳数据,如果您的峰非常拥挤(例如在 TP-PCR 或等位基因 ladder 中),您获得的峰面积可能与真实值不同。在这种情况下,您必须使用基线校正和去噪选项。
FragalyseQt 不会更改所分析的文件,因此如果您想保存分析结果,必须将其导出为 CSV 格式。
## FragalyseQt 快捷键。
`Ctrl+O` — 打开文件进行分析。
`Ctrl+E` — 将分析结果导出为 CSV。
`Ctrl+I` — 导出内部分析数据为 CSV(仅限 ABI 3500 和 SeqStudio 系列)。
`Ctrl+Shift+C` — 导出为 CODIS 3.2 CMF XML。
`Ctrl+Shift+A` — 添加自定义分子量内标。
`Ctrl+W` — 关闭当前标签页。
`F1` — 关于。
## FragalyseQt 计划添加哪些功能?
目前,我计划进一步改进分箱功能、基因分型支持以及 CODIS 导出覆盖范围。
## 我该如何支持您?
您可以提供来自我尚未测试过的机器的运行文件,提供补丁,或在资金上支持我。无论是哪种情况,都可以通过电子邮件 ( dorif11 AT gmail DOT com )、Telegram ( t.me/nurgle ) 或在 GitHub 上联系我。每一份帮助都非常重要!
## FragalyseQt 采用什么许可证?
0.4.x 之前的版本采用 GNU GPL v3。0.4 及以上版本采用 GNU AGPL v3。
## 从预编译包安装
预编译的 DEB 和 RPM 包可在
[发布页面](https://github.com/Dorif/fragalyseqt/releases)获取。
### DEB (Debian, Ubuntu, Linux Mint 及其衍生版)
下载 `.deb` 文件并使用 `apt` 安装:
```
sudo apt install ./fragalyseqt_0.5.1_all.deb
```
这将自动解析并安装所有依赖项。
### RPM (Fedora, openSUSE, RHEL 9+, Alma, Rocky 及其衍生版)
下载 `.rpm` 文件并使用 `dnf` 安装:
```
sudo dnf install ./fragalyseqt-0.5.1-1.noarch.rpm
```
在使用 `yum` 的旧版 RHEL/CentOS 7-8 系统上:
```
sudo yum install ./fragalyseqt-0.5.1-1.noarch.rpm
```
## 如何在 *ubuntu/Debian/SUSE/macOS 上运行?
以您喜欢的任何方式安装 Python 3.8+、git 和 PyQt5/PyQt6/PySide6(仅支持这些 Python 的 Qt 绑定)。之后,从 Git 仓库克隆 FragalyseQt,安装并运行它。我建议您使用 pip3 执行此操作:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip3 install . && fragalyseqt
```
如果遇到 externally-managed-environment 错误,请尝试:
```
pip3 install --break-system-packages .
```
在正常情况下,这实际上不会破坏任何东西。
或者,您可以尝试设置虚拟环境或使用 pipx,但前面提到的选项是推荐的。
如果您正在使用基于 RHEL 的系统 - 请阅读下一章,祝您好运。
## 如何在 AltLinux 上运行?
准备运行 FragalyseQt 的环境(需要 GCC 和 python3-dev 来构建 BioPython):
```
su -c "apt-get install -y git gcc python3-module-pip python3-module-scipy python3-module-numpy-testing python3-dev"
```
下载、安装并运行 FragalyseQt:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip install . && fragalyseqt
```
## 如何在 RHEL/Alma/Rocky/Oracle 8+ 上运行?
准备运行 FragalyseQt 的环境:
```
sudo yum install python-qt5 git
```
下载、安装并运行 FragalyseQt:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip install . && fragalyseqt
```
## 如何在 RHEL/CentOS/Alma/Rocky/Oracle/Scientific 7 上运行?
这些系统的软件仓库中没有 PyQt5,您也无法在全新安装的这些系统上安装 PyQt5。要安装 PyQt5,您应该首先更新 pip3 并清理 pip 缓存:
```
sudo pip3 install --upgrade pip && rm -rf .pip/cache
```
之后安装 FragalyseQt 运行所需的包:
```
pip3 install pyqt5 && sudo yum install git
```
下载、安装并运行 FragalyseQt:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip install . && fragalyseqt
```
## 如何在 FreeBSD 上运行?
首先,安装 Git 和运行 FragalyseQt 所需的库(假设您已经安装了 Python 3.9):
```
pkg install git graphics/py-pyqtgraph biology/py-biopython textproc/py-charset-normalizer py39-pip
```
现在,下载 FragalyseQt,安装并运行它:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip-3.9 install . && fragalyseqt
```
或者,如果您希望从 PIP 安装所有的 python 库(但请确保它们没有作为原生包安装):
```
pkg install git py39-pip && git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip-3.9 install . && fragalyseqt
```
## 如何在 OpenBSD 上运行?
首先,安装 Git 和运行 FragalyseQt 所需的库:
```
pkg_add git py3-pip py3-qt5 py3-pyqtgraph py3-biopython py3-charset-normalizer py3-scipy
```
现在,下载 FragalyseQt,安装并运行它:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip3 install . && fragalyseqt
```
或者,如果您希望从 PIP 安装所有的 python 库(但请确保它们没有作为原生包安装):
```
pkg_add git py3-pip && git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip3 install . && fragalyseqt
```
## 如何在 NetBSD 上运行?
首先,安装 Git 和运行 FragalyseQt 所需的库。在这种情况下,通过 pip 安装 pyqtgraph 是至关重要的,因为原生的 NetBSD 包似乎在某些方面存在问题(假设您使用的是相当新版本的 NetBSD 和 python 3.11):
```
pkg_add git py311-pip py311-qt5 py311-biopython py311-charset-normalizer py311-scipy && pip3.11 install pyqtgraph
```
现在,下载 FragalyseQt,安装并运行它:
```
git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip3.11 install . && fragalyseqt
```
或者,如果您希望从 PIP 安装所有的 python 库(但请确保它们没有作为原生包安装):
```
pkg_add git py311-pip && git clone https://github.com/Dorif/fragalyseqt && cd fragalyseqt && pip3.11 install . && fragalyseqt
```
## 它能在非 x86 机器上运行吗?
是的,可以,因为它不仅在操作系统方面是跨平台的,在硬件方面也是跨平台的。它至少已在 RISC-V 和 ARM 架构的机器上运行过。
## 已知存在问题的系统。
OpenVMS - 没有 Qt,至少在 x86_64 版本上没有稳定的 Python,不用考虑了。
Solaris/Illumos - 无法构建和安装 SciPy。
Haiku - pyqtgraph 包似乎存在问题(通过 pip 安装可解决),SciPy 包编译时缺少 ndimage 模块且通过 pip 安装会失败,也许将来会修复这个问题。
## 如何进一步提升 FragalyseQt 的速度?
如果您使用的是 Linux 或 macOS,您可以为此安装 Pyston(一个针对 Python 的性能优化 JIT),它将加速兼容 Python 3.7 - 3.10 的应用程序,不仅限于 FragalyseQt:
```
pip3 install pyston_lite_autoload
```
## 如何选择用于 sizing 的分子量内标?
这可能有些违背直觉,但有时您必须使用与在 CE 数据图正上方检测并显示的内标不同的分子量内标。
以下是一些关于如何选择正确分子量内标的提示:
如果您看到数据图上方写有 LIZ/CC5/WEN/BTO/其他通道 5 的 ladder,但没有可用的 LIZ/CC5/WEN/BTO 通道数据 - 请根据您的试剂盒和设备规格选择 ROX/CXR/其他通道 4 的 ladder。示例:Profiler Plus、CoFiler 和 PowerPlex 16 的所有样本文件。
如果您看到有限范围的 ladder 子集(如 GS600LIZ(60-460)),但看到的峰明显大于该 ladder 的上限 - 请计算峰的数量,并在已计数的峰中选择具有最高峰大小的 ladder。示例:在作为示例提供的 SeqStudio GlobalFiler 文件中,检测到了 GS600LIZ(60-460) ladder,但实际上存在高达 600 个核苷酸的所有 ladder 峰,因此您必须使用 GS600LIZ、GS600LIZ(60-600)、GS600LIZ(80-600) 或 GS600LIZ(100-600) ladder 进行 sizing。
如果在该低大小区域有影响甚至 ladder 通道的强烈噪音,请选择跳过低大小标记的 ladder 子集,如 GS600LIZ(60-600)。
仅当相应的峰未能成功进样时,才选择带有减号峰的 ladder 子集,例如 GS600LIZ-250。
## 如何选择 sizing 算法并对我的数据进行 SizeCall?
速度最快且消耗资源最少、同时具备中等精度的是 Linear Spline。如果您使用的是极度弱势的系统,例如旧款上网本或第一代 Raspberry Pi,请选择它。
最均衡的是 Cubic Spline。
5 次样条曲线可能比 Cubic 稍微精确一些。
然而,Cubic 和 5 次样条曲线都存在 Runge 现象,这使得它们在处理极小或极大的尺寸时几乎无法使用。
如果您希望更高的 sizing 均匀性 - 请使用 LSQ 加权样条 sizing。其精度从 1 阶到 5 阶逐渐提高。资源消耗也会以同样的方式增加。
纯 LSQ calling 擅长处理具有某种程度异常迁移的高度多重化和嘈杂数据。
每次您更改峰检测设置、应用基线校正和去噪、隐藏/显示通道或打开新文件时,请点击 "SizeCall" 按钮。
## 关于测试文件的来源。
来自 ABI 3100 的 FSA 文件和来自 ABI 3500 的 HID 文件来自 NCBI OSIRIS 测试文件集。NCBI OSIRIS 是公共领域软件。
NCBI OSIRIS 源代码获取地址:https://github.com/ncbi/osiris
NCBI OSIRIS 主页:https://www.ncbi.nlm.nih.gov/osiris/
来自 ABI 310 的文件来自 MLPAinter 项目,采用 GNU GPL v3 许可。
MLPAinter 源代码获取地址:https://code.google.com/archive/p/mlpainter/
MLPAinter 出版物全文获取地址:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-67
NIST STR 文件来自 NIST Forensic DNA Open Dataset v. 1.3.0,也可在此获取:https://data.nist.gov/od/id/mds2-2157/pdr:v/1.3.0
NIST mixtures STR 文件来自 NIST Forensic DNA Open Dataset v. 1.4.1,也可在此获取:https://data.nist.gov/od/id/mds2-2157/pdr:v/1.4.1
根据其他 NIST 数据/作品的合理使用政策,已在相应目录中添加了版权声明:
https://www.nist.gov/open/copyright-fair-use-and-licensing-statements-srd-data-software-and-technical-series-publications
这些数据集中附带的 Panel 文件已在单独的目录中,以避免文件重复。
用于 GlobalFiler 的 SeqStudio 和 RapidHIT ID v1.x 文件由本人获取,不包含任何其他人的数据。
用于 Nanophore-05 和 SeqStudio 的 COrDis Plus 文件由莫斯科 ISB 的 Dmitrii Romaniuk 提供,不包含个人数据。
Superyears Honor 1616 文件由俄罗斯别尔茨克 "Litolab" 的 Vadim Ponkratov 提供,不包含个人数据。
## 这是什么?
这是一款用于片段分析(MLPA、QF-PCR 等)数据处理的软件。
## 功能特性
FragalyseQt 0.4 及以上版本支持同时使用多达 8 种不同染料的检测,支持选择性通道隐藏、非拉丁语运行名称,能够正确处理损坏的文件,支持为任何 FSA 或 HID 文件以 CSV 格式导出峰位置、面积、FWHM、大小和通道名称,并支持将 ABI 3500 和 SeqStudio 系列设备生成的 FSA 或 HID 文件的内部分析数据导出为 CSV 格式。自 0.4.2 版本起,FragalyseQt 还支持 Global 和 Local Southern sizing(如 doi: 10.1016/0003-2697(79)90235-5 中所述)。峰面积的计算假设其符合高斯分布。对于噪声较大的运行,提供了去噪和基线校正选项。
自 0.5 版本起,支持使用来自 GeneMapper(制表符分隔的 `*_Panel*.txt`,可附带 `*_Bins*.txt` 和 `*_Stutter*.txt`)和 GeneMarker (`.xml`) 的 panel 文件进行等位基因分箱。**NCBI OSIRIS LadderInfo XML** panel 文件(`.xml`,根元素为 `标签:ABI 3500, CMF XML, CODIS 3.2, DNA片段分析, FSA文件处理, GeneMapper, GeneMarker, HID文件解析, MLPA, OSIRIS, QF-PCR, SeqStudio, Southern算法, STR分析, Stutter过滤, 基因分型, 基线校正, 实验数据管理, 峰值导出, 数据分析工具, 法医数据库, 法医遗传学, 生物信息学, 等位基因分箱, 逆向工具, 降噪, 高斯拟合